
The European Union Medical Device Regulation (EU MDR) has brought significant changes to the compliance timeline. With the transition timeline extension, manufacturers have an opportunity to ensure compliance with the new regulations. However, it’s important to understand the conditional nature of the extension and the requirements that must be met. In this blog, we will provide you with five essential tips to navigate the EU MDR extension successfully.
Evaluate your product portfolio: To determine if you are eligible for the MDR transition timeline extension, it’s crucial to evaluate your product portfolio. The EU Commission has published a helpful guidance document that can assist you in this process. You can find the document here. It’s important to carefully review the guidance and assess if your products meet the criteria for the extension.
Ensure comprehensive NB application: When submitting your application to the Notified Body (NB), make sure it includes all the necessary information. Common pitfalls to avoid include gaps or inconsistencies in device description, intended purpose, device classification, and justification. Additionally, provide accurate information about critical subcontractors, their roles, and the current state of their QMS certification. Pay attention to marketing claims that may impact the need for a clinical evaluation consultation procedure.
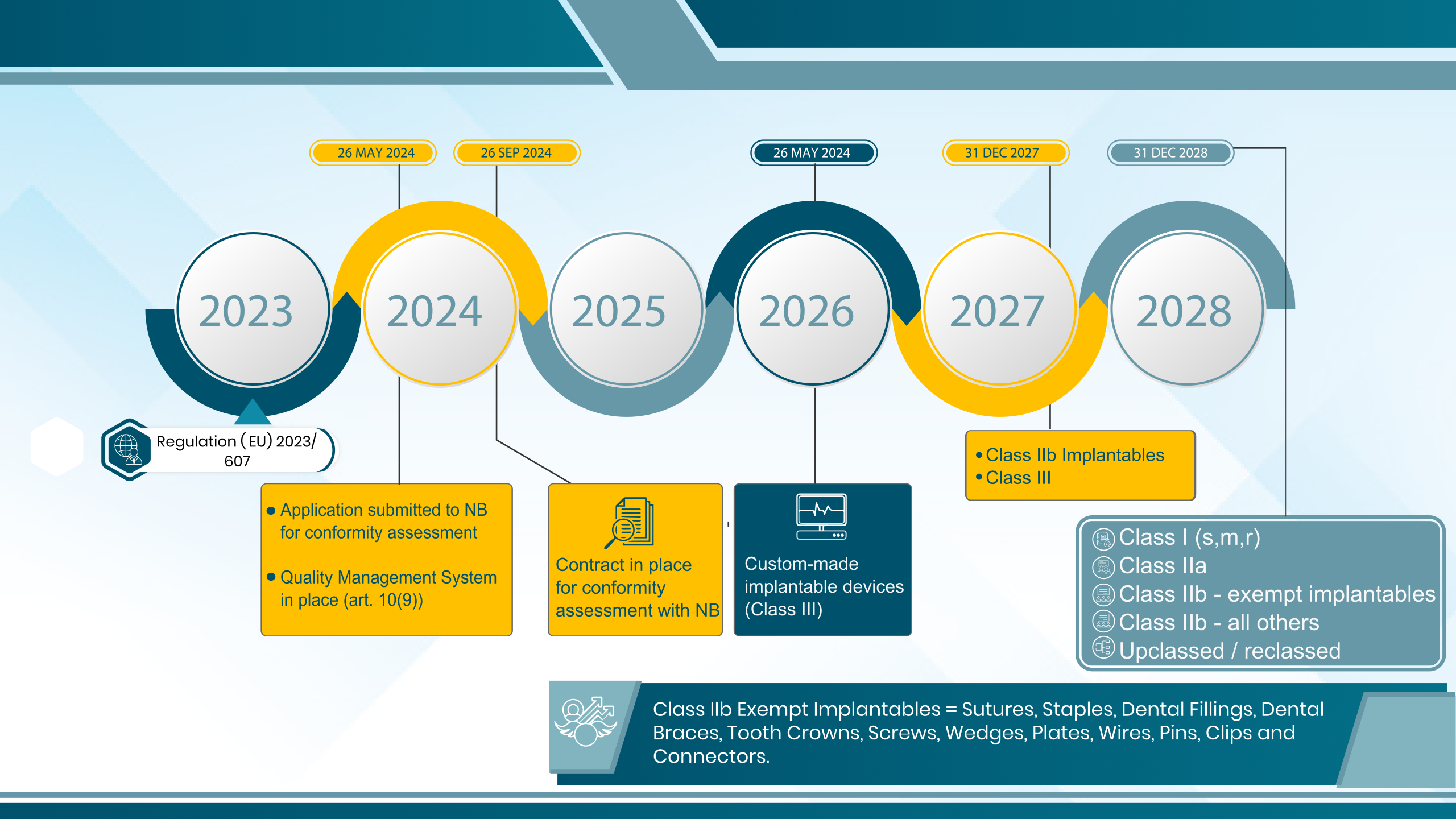
Meet the interim deadlines: To benefit from the MDR transition timeline extension, manufacturers must meet two critical deadlines. By 26 May 2024, manufacturers must have lodged an MDR application with their NB. Subsequently, by 26 September 2024, the NB must have accepted the application. These interim deadlines are in place to prevent a last-minute rush of applications as the new deadlines approach. It’s crucial to adhere to these deadlines to ensure a smoother transition process.
Other requirements are:
- Without substantial modifications to their design or intended purpose, the devices must conform to Directive 90/385/EEC (AIMDD) or Directive 93/42/EEC (MDD).
- The device must not pose any unacceptable hazards to the health or safety of patients, users, or other individuals or any other component of public health protection.
- The manufacturer must have a Quality Management System (QMS) in place, conforming with Art 10(9) by May 26, 2024, at the latest.
- Since May 26, 2021, all actors and devices must be registered, and the maker or authorized representative must comply with MDR Article 120 criteria for Post-Market Surveillance (PMS), vigilance, trend reporting, market surveillance, and registration.
Work with NBs: By submitting your applications early, you enable NBs to effectively plan their certification workload. This collaboration ensures a more streamlined transition process and reduces the risk of delays. Work closely with your NB to address any queries or concerns they may have during the assessment process.
Conclusion: Navigating the EU MDR transition timeline extension requires careful evaluation, comprehensive applications, and adherence to interim deadlines. By following these five essential tips, manufacturers can master the transition and achieve EU MDR compliance successfully. Remember, staying proactive and informed is key to ensuring a smooth transition to the new regulations.